METODOLOGíA DE LOS ENSAYOS CLíNICOS EN EL ASMA
Valentina Gutiérrez Vall de
Cabres
Hospital Universitario Dr
Peset. Valencia
Muchos de los tratamientos que empleamos
habitualmente siguen siendo objeto de controversia, en gran parte por falta de
evidencias sólidas sobre su efectividad. Se gastan cantidades ingentes de
recursos en intentos terapéuticos (o diagnósticos) de valor incierto. En estas
circunstancias, los ensayos clínicos bien diseñados podrían representar ahorro.
Las “ideas” más o
menos prometedoras que podrían dar base a una buena alternativa de tratamiento
suelen inferirse a partir de los mecanismos etiopatogénicos de la enfermedad, o
surgen de apreciaciones de la clínica o de estudios epidemiológicos. Así, el
reconocimiento del asma como enfermedad inflamatoria ha dado soporte científico
al tratamiento con esteroides inhalados, y la apreciación de cambios
persistentes en la pared de las vías aéreas ha instado a investigar acerca de
la necesidad de la adición de otro fármaco antiasmático en muchos casos.
Pero en
investigación, las apariencias engañan, nada puede darse por sentado y e sentido común, tan necesario, no basta.
La mejor forma de someter estas ideas a examen parece ser el ensayo controlado, aleatorizado, simultáneo
y ciego (1) que trata de un tipo especial de estudio de cohortes en el que la
intervención terapéutica se asigna minimizando los sesgos.
Los ensayos clínicos
están ética y científicamente justificados por varias razones:
- En muchas
enfermedades, los tratamientos disponibles son insuficientes o claramente
ineficaces, por lo que es necesario encontrar mejores tratamientos.
- La experimentación animal tiene bajo poder
predictivo para la generalización de los hallazgos a las personas.
- En muchos casos no se dispone de
alternativas científicas para establecer el verdadero valor terapéutico de una
nueva sustancia.
- Del ensayo clínico, pues, derivan múltiples
ventajas para el enfermo, la sociedad, el médico, el hospital y la ciencia en
general.
Por
otro lado, cuando los ensayos clínicos se incorporan, como una actividad
asistencial más, a una estructura clínica hospitalaria, se aportan mejoras no
solo en la calidad asistencial y en el desarrollo científico, sino también en
la organización del trabajo y en el fomento del estudio.
PRINCIPIOS
DE LA INVESTIGACIóN CLíNICA APLICADA.
Aunque no todos los autores
están de acuerdo, parece claro que existe una ciencia de la investigación
clínica aplicada, que funciona. La prueba no deja lugar a dudas: la medicina moderna
es efectiva para prevenir y tratar muchas enfermedades. Pero ¿en qué consiste
esa ciencia y cómo funciona?. Determinar cuál es la mejor forma de hacer
investigación sobre los problemas clínicos no es una tarea fácil, y no existe
ningún planteamiento que se considere unánimemente como el mejor. Quizás el más
importante sea esforzarse en el ideal de
ser perfectamente sistemático. La
realidad práctica nos hará descubrir en qué situaciones no se puede evitar
apartarse de estos ideales.
¿Qué es lo más importante en investigación
clínica?
Quizás las tareas más importantes sean las
siguientes:
- Ser
creativo, y detectar las preguntas importantes sobre las cuales investigar,
e invertir tiempo y dinero, y diseñar planteamientos ingeniosos para obtener la
respuesta correcta.
- Tener
buen criterio para saber combinar los objetivos científicos y prácticos que
compiten entre sí, desafiando la pericia de quien construye el estudio. Por
todas partes aparecen alternativas difíciles de decidir.
- Un buen investigador se distingue por su sentido común al formular la
pregunta en que se basará la investigación, y al escoger un diseño en
particular, determinando la forma concreta en que se obtendrán los individuos
del estudio, el tamaño de la muestra y las mediciones que se efectuarán
VISIóN
GLOBAL DEL PROCESO DE INVESTIGACIóN CLíNICA
La
“obra” comienza con la piedra angular filosófica de la investigación: el
proceso de hacer inferencias sobre la
verdad en la situación real a partir de
los hechos observados en la muestra que se estudia y finaliza con la
elaboración de una memoria a fin de obtener la financiación económica para la
investigación. Entre ambas cosas, está el difícil entramado de diseñar y llevar
a cabo un proceso de investigación, que puede dividirse en seis etapas (Figura
1).

La
primera etapa es la elección de la pregunta a investigar, sobre un problema
concreto de salud en el mundo real, que el investigador quiere contestar
realizando el estudio. Con frecuencia, la elección de una buena pregunta a investigar es el mayor desafío al que debe
enfrentarse un nuevo investigador. Elegir un buen director con experiencia y
buen criterio es importante en estos casos.
El
segundo paso consiste en desarrollar el protocolo del estudio. El objetivo es
diseñar un protocolo barato y factible en cuanto a su realización, que
proporcione una respuesta correcta a la pregunta que se investiga. Para ello es
necesario decidir cómo reclutar a los
individuos que formarán parte del estudio, elegir la forma de realizar las determinaciones
y el diseño el estudio, estimar el tamaño de muestra, enfocar los aspectos
éticos, y planificar la forma de manejar y analizar los datos.
Después
se impone la revisión del protocolo, y estudio piloto si procede, y la fase de ejecución del mismo, que suele ser
la más costosa en cuanto a tiempo y dinero.
Por
último, en lo relativo al análisis de los hallazgos y a la obtención y difusión
de las conclusiones, es fundamental aprender a extraer las conclusiones
correctas a partir de la investigación y decidir de qué modo pueden aplicarse a
las decisiones clínicas.
Los
últimos pasos llevan de manera natural al planteamiento del siguiente proyecto.
La mayoría de estudios generan más preguntas de las que contestan, y muchos
investigadores se dan cuenta de que a medida que adquieren experiencia van
descubriendo más problemas que podrían investigarse.
Además,
con la experiencia, la investigación se hace más fácil y gratificante. Nos
vamos familiarizando con los pormenores del reclutamiento de pacientes, con la
medición de variables, con los diseños, etc. Cada vez las solicitudes de
ayuda llegan a buen fin con menos dificultades, se va reuniendo un
grupo de personal colaborador e
investigadores más jóvenes y se establecen contactos gratificantes con científicos
que trabajan en los mismos temas realizándose a veces, con el esfuerzo
conjunto, avances interesantes en el conocimiento.
ESTUDIOS
EXPERIMENTALES. EL ENSAYO CLíNICO
El ensayo clínico es
un estudio experimental que se realiza en enfermos o en individuos sanos con el
fin de evaluar la eficacia o la
seguridad de un fármaco o un procedimiento terapéutico. Es el paradigma del
método científico aplicado a las ciencias de la salud y durante los últimos
años su número ha aumentado enormemente debido a que existe un mayor número de
fármacos disponibles en el mercado, y a los requerimientos, cada vez más
rigurosos, por parte de las autoridades
sanitarias para demostrar la eficacia de los fármacos que se han de comercializar.
El Real Decreto
561/93, define al ensayo clínico como “toda evaluación experimental de una
sustancia o medicamento a través de su aplicación a seres humanos. Sus fines
pueden ser múltiples: estudios de farmacocinética o farmacodinámica, establecer
la eficacia para una indicación, valorar los efectos profilácticos, investigar
su seguridad o conocer el perfil de reacciones adversas.
Según sus objetivos
y el momento del desarrollo del fármaco
se clasifican en:
Los ENSAYOS CLíNICOS DE FASE I son estudios
de farmacocinética (que evalúan la absorción, distribución, metabolismo y
excreción) o de farmacodinámica (que valoran las interacciones entre el fármaco
y el receptor). Es decir, proporcionan
información preliminar sobre el efecto y la seguridad de una sustancia en individuos sanos o en pequeños grupos de
enfermos, orientando sobre la pauta de administración mas adecuada para ensayos
posteriores. Constituyen el primer paso en la investigación de un nuevo
medicamento en seres humanos y suceden a los estudios preclínicos llevados a cabo
en animales de experimentación.
Los ENSAYOS
DE FASE II se realizan en enfermos que padecen el proceso patológico de
interés y tienen como fin proporcionar información preliminar sobre la eficacia
de una sustancia, al establecer la relación entre la dosis pautada y la
respuesta obtenida. Amplían los datos de seguridad y eficacia obtenidos en la
fase I.
ENSAYOS
CLíNICOS DE FASE III: Están destinados a evaluar la eficacia y la
seguridad de un tratamiento que todavía se considera experimental. Se trata de
reproducir las condiciones de uso habituales, considerando además las
alternativas terapéuticas disponibles en esa indicación.
Los ENSAYOS
CLíNICOS DE FASE IV se realizan con medicamentos ya comercializados y
suelen estudiar aspectos aún no valorados o en condiciones de uso distintas a
las autorizadas, como nuevas indicaciones. Sus objetivos son variados y la
investigación se plantea en condiciones similares a la práctica clínica
habitual. Suelen incluir gran número de pacientes y en ellos se presta especial
atención a los efectos adversos.
FRACASO
DE UN ENSAYO CLíNICO
Los ensayos clínicos no
siempre tienen éxito en su desarrollo y realización. Pueden fracasar bien
porque el producto carezca de la
eficacia que se estimaba o porque presente una toxicidad inaceptable. Sin
embargo, la mayor parte de las veces, los ensayos clínicos fracasan porque no
se alcanzan los objetivos, casi siempre por reclutamiento insuficiente de
participantes, que suele representar pérdida significativa del poder
estadístico.
Los ensayos clínicos
conllevan un enorme esfuerzo económico, gran dedicación de tiempo y una
mentalidad rigurosa y abierta de los investigadores. Por ello, participar en un
ensayo clínico tiene implicaciones personales, hospitalarias y científicas.
Además, la investigación clínica está sujeta a normas éticas y morales y existe
una regulación específica de cada país cuya finalidad es siempre que el proyecto beneficie al enfermo y a la sociedad
bajo la premisa de que la investigación sea humanitaria y útil desde el punto
de vista científico.
Estos aspectos se
han ido plasmando a lo largo de los años en diversos documentos y códigos
internacionales. Entre otros:
-Nuremberg 1974
-Declaración de
Helsinki de 1964
- Directiva de la
Comunidad Económica Europea sobre medicamentos de 1965
- Declaración de la
Organización Mundial de la salud de 1966 con sus revisiones (Tokio 1975,
Venecia 1983, Hong Kong 1989)
- Informe de
Belmonte 1978
- En España: Real
Decreto sobre Ensayos clínicos 1993
- Normas de Buena
Práctica Clínica
- Regulación del
Documento de Consentimiento Informado
Los
requisitos metodológicos de un ensayo clínico son múltiples y rigurosos. Antes
de comenzar deben quedar establecidas y bien definidas numerosas
circunstancias. En el asma, es obligado precisar y delimitar adecuadamente
diversos aspectos propios y específicos de esta enfermedad: ¿cómo se define la
enfermedad (datos clínicos y de exploración funcional)?, ¿cómo se excluyen otros procesos?, ¿qué tipo
de asma (ocupacional, polínica, de esfuerzo, etc)? ¿qué medicación antiasmática
y concomitante se permite?. Deben también precisrse las variables primarias y
secundarias de eficacia, las características y los patrones de calidad que deben cumplir los procedimientos
técnicos y los equipos de medida empleados así como el método a emplear para
comprobar el cumplimiento del protocolo y la adherencia a la pauta de
medicación empleada. Por último, ha de decidirse cómo van a evaluarse y
notificarse los efectos adversos y los criterios de salida o interrupción del
ensayo.
ENSAYOS CLíNICOS, EXPERIMENTOS O ESTUDIOS
EXPERIMENTALES
El
objetivo de los estudios experimentales es evaluar la eficacia de cualquier
intervención preventiva, curativa o rehabilitadora. Se definen por dos
características. La primera es que los investigadores tienen control sobre el factor de estudio, es decir, deciden qué
tratamiento, con qué pauta, y durante
cuánto tiempo recibirá cada uno de los grupos del estudio.
La segunda característica es
que la asignación de los individuos a los grupos del estudio se realiza de
forma aleatoria. Si los grupos así obtenidos
son comparables, cualquier diferencia observada entre ellos al final del
experimento puede ser atribuida, con bastante convicción a la diferente
intervención a la que han sido sometidos. La inferencia de causalidad se basa
en a comparación de los desenlaces observados en individuos clasificados según
la intervención que recibieron (una forma de variable predictiva).
La gran ventaja del ensayo clínico es su
alto grado de control de la situación que proporciona, en el caso de que
exista una relación entre la intervención y la respuesta observada, una fuerte
inferencia de causalidad.
Los ensayos clínicos son
idóneos para poner a prueba la eficacia de estrategias de tratamiento y representan
el mejor diseño para controlar la influencia de variables de confusión.
Por otro lado, los ensayos
clínicos suelen reservarse para preguntas a investigar relativamente “maduras”:
en enfermedades para las cuales los estudios observacionales hayan revelado las
características descriptivas básicas, la etiología sugerida, y se haya señalado
un camino para enfocar el problema desde un punto de vista clínico.
En los diseños entre grupos se comparan los desenlaces observados en dos
(o más) grupos de individuos que reciben diferentes intervenciones.
En los diseños intra-grupo se comparan los desenlaces observados en un
solo grupo, antes y después de aplicar una intervención. Los primeros son los más extendidos en
investigación clínica. Uno de ellos, el
ensayo clínico aleatorio y ciego se suele presentar como el estándar óptimo o ideal, y es el que desarrollaremos
en este capítulo.
ESTRUCTURA DE UN ENSAYO CLíNICO ALEATORIO
El ensayo clínicos aleatorio
es el estudio experimental más frecuente en nuestro ámbito. Abordan preguntas
como: ¿el contacto precoz con mascotas previene la sensibilización alérgica?,
¿El tratamiento con inmunoterapia previene la evolución a asma en sujetos con
rinitis alérgica? o ¿la adición de un segundo fármaco a los corticoides inhalados
permite reducir sus dosis?. Los pasos de un ensayo clínico aleatorio se
esquematizan en la figura 2.
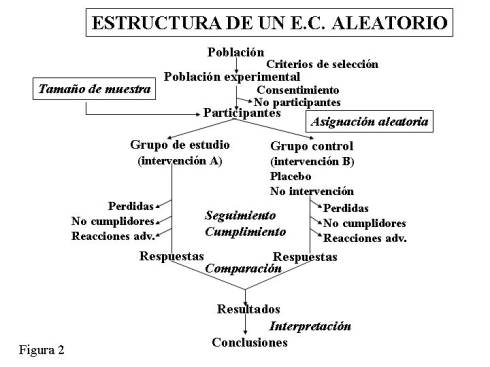
1.
Selección de la población
Los individuos en los que se
lleva a cabo la intervención proceden de una población de referencia o diana a la que se quieren extrapolar los
resultados. Su selección responde a las preguntas: ¿qué tipo de sujetos se
estudiarán? ¿cómo se reclutarán?
A partir de esta población
diana, se especificarán unos criterios de selección que darán lugar a una población de estudio o muestra que es
aquella en la que se desea realizar la experiencia.
Los criterios de inclusión han de definir los sujetos que
potencialmente se pueden beneficiar de
la intervención. Si estos criterios son muy amplios, la muestra será más
representativa de la población de referencia y las posibilidades de generalizar
los resultados son mayores. Sin embargo, en una muestra muy heterogénea será
más difícil hallar la respuesta esperada y se requerirá un mayor número de
individuos.
Hay que incluir a los
pacientes en los que la intervención está contraindicada y a los que se supone que no van a cumplir las
indicaciones del protocolo de estudio, así como aquellos con trastornos
extraños que competirán con el desenlace. Esto limita la generalización de los
resultados pero permitirá recoger una información más fiable sobre la que basar
la interpretación.
Así pues, la utilización de
criterios de inclusión y exclusión estrictos aleja a la población de estudio de
la población diana, y por lo tanto va en contra de la generalización, pero
aumenta la validez de las observaciones.
Antes de que los individuos
de la población experimental lleguen a entrar en el estudio deben dar su consentimiento, manifestar que aceptan
participar. Es muy probable que aquellos que los dan difieran en múltiples
aspectos de los que no lo hacen, incluyendo la motivación y actitudes hacia la
salud y factores de riesgo de la enfermedad. Esto dificulta, también, la
generalización de los resultados. De todos modos, está ampliamente aceptado que
la participación en un ensayo clínico aleatorio debe ser voluntaria y basada en
el consentimiento informado.
2.
Estimación del tamaño de muestra
Los experimentos con un número demasiado pequeño de individuos
para detectar efectos significativos en la población han sido muy frecuentes en
el pasado, desafortunadamente. La estimación de las necesidades del tamaño de
muestra es una de las partes más importantes de la planificación de un estudio.
Cuanto
mayor es la muestra menos probable es que variables (conocidas o no) que pueden
influir en el resultado se distribuyan de modo desigual entre los grupos y, en
consecuencia, es poco probable que la respuesta esperada varíe a no ser como
resultado del tratamiento aplicado.
A
veces el reclutamiento es difícil y debe planificarse que la población sea suficiente, accesible, y se disponga
del tiempo y el dinero suficiente para obtenerla.
3. Asignación Aleatoria
Una
vez se ha elegido la población, el siguiente paso es asignar a los individuos a cada uno de los grupos (Figura 2).
En los ensayos clínicos aleatorios, se lleva a cabo de forma randomizada. Un
ensayo clínico aleatorio admite muchas variaciones en su diseño: pueden ser
estudios realizados en un solo centro o bien multicéntricos, pueden utilizar o
no técnicas de enmascaramiento o comparar un tratamiento con otro o con
placebo, según las circunstancias, pero la condición sine qua non para definirlo es la asignación aleatoria de los
participantes a los grupos de estudio.
Los
beneficios que se obtienen de la asignación aleatoria se pueden resumir en dos
puntos (2):
- Se consigue que
la comparación entre ambas intervenciones o tratamientos sea lo más
imparcial posible. Cuanto mayor es la muestra de sujetos
estudiada, menor es la probabilidad de que las variables, conocidas o no,
que pueden influir en el resultado (edad, sexo, características basales),
se distribuyan de modo desigual entre los grupos, y por ello será también
poco probable que la respuesta esperada (criterio de evaluación) varíe
ampliamente a menos que sea, efectivamente, resultado del tratamiento
aplicado. La estadística permitirá dar una estimación cuantitativa del
término poco probable. Cuando se estudian pocos pacientes pueden existir
importantes diferencias, a pesar de que los sujetos se hayan asignado aleatoriamente.
La asignación aleatoria no garantiza que los grupos sean idénticos, sin
que simplemente aumenta la probabilidad de que los sean.
- La segunda ventaja es que la
asignación al azar permite la utilización
de técnicas de enmascaramiento, que son muy útiles para obtener
una estimación no sesgada del criterio de evaluación.
La asignación aleatoria es
la clave de un ensayo clínico aleatorio por lo que debe realizarse
correctamente, de modo que ni las preferencias del médico ni las del paciente
influyan en la decisión de a qué grupo se asigna al participante.
- Técnicas de
enmascaramiento.
Las expectativas, tanto de los pacientes como de los médicos, pueden
influir en la evaluación de la respuesta observada. Este problema se evita
utilizando las llamadas técnicas de ciego
o de enmascaramiento que se definen como aquellos procedimientos realizados
con el fin de que los miembros del equipo investigador y/o los participantes
del estudio no conozcan algunos hechos u observaciones que pudieran influir en
sus acciones o decisiones y sesgar los resultados.
VENTAJAS
Un investigador puede no ser completamente imparcial en la evaluación
de dos o más intervenciones. Siempre existe un cierto grado e escepticismo o
prejuicio hacia una de ellas. Incluso si es completamente imparcial, los
primeros resultados pueden influenciar sus expectativas creando un cierto
entusiasmo o desilusión que será difícil de ignorar, introduciendo un sesgo en
la interpretación de los resultados. Si los investigadores conocen quién recibe
la intervención existe la posibilidad de que, inconscientemente, se examine con
mayor minuciosidad cualquier respuesta o se pregunte con más detalle sobre
efectos secundarios. Son las intervenciones
no intencionadas o cointervenciones, que pueden influir en las diferencias.
Estas preferencias también se dan en los pacientes. El investigador
puede contagiar su entusiasmo o pesimismo sobre la nueva intervención a los
participantes del estudio, influenciado la respuesta. Por todo ello es
importante la utilización de técnicas de enmascaramiento siempre que sea
posible, y muy especialmente cuando la respuesta sea difícil de medir porque
interviene la subjetividad del investigador o del paciente, como ocurre con la
disnea o las puntuaciones de síntomas, fácilmente influenciables por el mero
hecho de estar bajo tratamiento.
Si bien la aleatorización
elimina la confusión debida a las variables basales, el enmascaramiento elimina
la confusión causada por las intervenciones no intencionadas.
LIMITACIONES
En los estudios farmacológicos, el enmascaramiento se consigue
presentando ambos fármacos con un formato idéntico. Sin embargo, el ciego se puede romper fácilmente si los
fármacos tienen una toxicidad o efectos secundarios distintos y bien conocidos,
como la faringitis en el caso de los corticoides inhalados o el temblor y
palpitaciones con los beta-adrenérgicos.
La aplicación de técnicas de ciego puede ser inviable cuando se evalúan
intervenciones no farmacológicas como las medidas de desalergenización o el
efecto de un posible agente etiológico.
- Placebo
El concepto de efecto placebo hace referencia al efecto
psicológico o fisiológico de cualquier medicación, independientemente de su
efecto farmacológico. Los factores que intervienen en el efecto placebo son la
propia personalidad del paciente, las convicciones y entusiasmo del equipo
investigador, y las condiciones de administración y características de la
intervención.
Por tratamiento placebo se
entiende una intervención de características fisicoquímicas (aspecto, sabor,
color, etc.) y pauta de administración similares a las del fármaco que se
evalúa, pero que no posee actividad farmacológica específica para la enfermedad
que se está estudiando.
VENTAJAS
La principal ventaja es la
de controlar los efectos derivados de cualquier característica del tratamiento
que no sea el efecto que se está estudiando. También permite la utilización de
técnicas de ciego cuando se evalúan dos fármacos con distinta presentación o
cuando la duración de los tratamientos es distinta.
Un placebo no solo tiene
efectos terapéuticos sino también efectos secundarios y reacciones adversas. Su
uso permite conocer la proporción de efectos indeseables que están causados en
realidad por la intervención.
LIMITACIONES
La primera y más importante es ética; siempre que exista un tratamiento que se ha mostrado eficaz
deberá ser empleado, no pudiéndose aplicar un tratamiento placebo. La segunda
es práctica ya que muchas
intervenciones no son susceptibles de ser comparadas con un placebo como las
recomendaciones ambientales o aspectos de educación sanitaria.
6.
Seguimiento
El principio que ha de guiar
el seguimiento de los participantes es el de asegurar la comparabilidad entre
los grupos, es decir, la pauta de
visitas y exploraciones ha de ser
idéntica para todos los participantes
El seguimiento debe ser adecuado a cada
problema concreto y lo suficientemente largo como para asegurar que se
producirá la respuesta esperada. En algunos casos será de pocas semanas, como
en las exacerbaciones asmáticas. En otros se prolongará años, especialmente
cuando se evalúan medidas de prevención primaria. Sin embargo, cuanto menor sea
el tiempo de seguimiento más facilidad existe para mantener el contacto con los
participantes y, por consiguiente, la probabilidad de pérdidas será menor. Las
fuentes de pérdidas durante el tiempo de observación son diversas y su número
suele estar en relación con la complejidad y la duración del protocolo.
Otra norma importante es que
la duración del seguimiento y las pruebas que se llevan a cabo han de ser
iguales para todos los participantes.
Por
último, cabe señalar que la posibilidad de que se produzca un sesgo no está en
relación con el número de pérdidas, sino que su respuesta a la intervención sea
distinta a la de los que finalizan el estudio.
7.
Cumplimiento
El
seguir un tratamiento no es tarea fácil, especialmente en las enfermedades
crónicas. Existen muchas oportunidades para que los pacientes dejen de cumplir
parte o todas las medidas que se les recomiendan.
La
tarea de los responsables del estudio es conseguir que exista el mayor
cumplimiento posible. La primera
estrategia para conseguirlo se relaciona con los criterios de inclusión,
eliminando a potenciales mal cumplidores. El hecho de que haya muchos malos cumplidores ya es un dato sobre la
efectividad del tratamiento
La
segunda estrategia es conseguir que
el porcentaje de mal cumplidores se distribuya por igual entre los grupos, lo
que se suele conseguir gracias a la asignación al azar.
La
tercera se relaciona con la organización el estudio. El
cumplimiento está en relación con la complejidad de la intervención y la
duración del seguimiento. Es conveniente explicar a los participantes la
importancia de que cumplan las medidas recomendadas y acudan a las citas que
determina el protocolo. Es una buena práctica dar las instrucciones de manera
concisa y por escrito.
El
mal cumplimiento tiene implicaciones en la interpretación de los resultados. En
primer lugar se puede comprometer la validez interna si se excluye a los no cumplidores del análisis. La adherencia a
un protocolo puede estar relacionada con el tratamiento, y si es mayor en uno
de los grupos, la exclusión de los malos cumplidores del análisis puede sesgar
los resultados.
7.
Aspectos éticos
A
diferencia de otros estudios, en los experimentales el investigador controla la
intervención, lo que plantea siempre dudas sobre la ética de su realización. El
propio término “experimentación humana” suscita recelos, a menudo. Sin embargo, no hay que olvidar que el
desarrollo de los ensayos clínicos en los años recientes ha representado un
gran avance porque cuando no está clara la eficacia de una intervención, un
ensayo clínico aleatorio bien diseñado y ejecutado es el tipo de estudio que
proporciona los resultados más fiables. En este sentido, sería una falta de
ética dar un tratamiento de eficacia no probada simplemente porque no se ha
llevado a cabo un ensayo clínico.
Se
pueden señalar las ss. restricciones
básicas:
1) Los factores de
estudio o exposiciones deben limitarse a los potencialmente preventivos de una
enfermedad o de sus consecuencias.
2) Las exposiciones en
todos los grupos de estudio deben ser igualmente aceptables según los
conocimientos actuales.
3) Los sujetos
incluidos en un estudio no deben ser
privados en ningún momento de las mejores medidas terapéuticas y preventivas.
Por ejemplo, no es ético utilizar un placebo en una situación para la que
exista un tratamiento efectivo.
4) Los sujetos deben
ser informados de su participación en un experimento y de sus posibles
consecuencias.
5) Restricciones
relativas a la protección de datos.
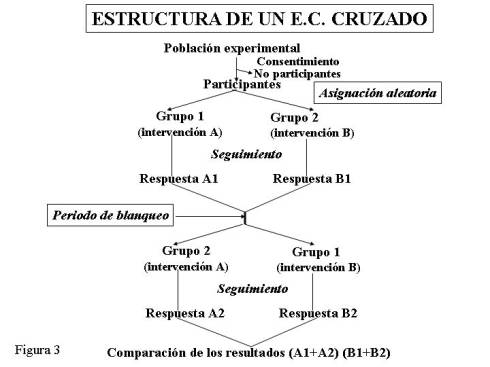
ENSAYO CLíNICO
CRUZADO
El tipo de ensayo clínico
anteriormente descrito corresponde a los estudios
en paralelo, donde cada paciente recibe solo una intervención y los
resultados obtenidos en cada uno de los grupos se comparan entre sí. Una forma
especial de ensayo clínico aleatorio, menos utilizada, es el diseño cruzado (cross-over). En el que
cada sujeto actúa como su propio control. En el caso más sencillo, cada
individuo recibe aleatoriamente una de
las dos intervenciones y, en un segundo
periodo, la otra. Ambos periodos están
separados por una fase de blanqueo, para permitir que el
paciente vuelva a su estadio inicial.
En un estudio farmacológico el periodo de blanqueo debe ser lo
suficientemente largo para asegurar que
el efecto del tratamiento administrado
en el primer periodo ha desaparecido. Las características diferenciales entre
un estudio en paralelo y uno cruzado se recogen en le Tabla 1.
Tabla 1. Principales
características diferenciales entre un estudio en paralelo y uno cruzado.
Estudio en paralelo
|
Estudio cruzado
|
|
-Cada participante recibe sólo una
intervención |
-Cada participante actúa como su propio
control |
|
-Cada participante se asigna a un grupo |
-Se asigna una secuencia de tratamientos a
cada participante |
|
-El periodo de blanqueo no es necesario |
-El periodo de blanqueo es imprescindible |
El diseño cruzado solo puede
utilizarse en enfermedades crónicas, relativamente estables, y en las que los
resultados de una intervención
desaparezcan de forma rápida. No
es aplicable en el caso de que el orden en que se administran las
intervenciones altere el resultado, o cuando no sea posible realizar un periodo
de blanqueo que asegure la total desaparición del efecto de la primera
intervención.
La duración del periodo de
blanqueo ha de ser corta. Un fármaco que necesita meses para ser eliminado del
organismo no es un buen candidato para ser evaluado en un estudio cruzado.
Igualmente, este diseño está contraindicado si el tiempo de blanqueo varía
significativamente de un individuo a otro. Estas ventajas y limitaciones se
resumen en la Tabla 2.
Tabla 2. Ventajas
y limitaciones de un estudio cruzado.
|
VENTAJAS -Es mas eficiente que un estudio en
paralelo, ya que requiere un número menor de participantes -Cada participante es su propio control,
por lo que se pueden utilizar técnicas estadísticas para datos apareados, más potentes. -LIMITACIONES -Tiene mayor duración que los estudios en
paralelo -No puede utilizarse en enfermedades agudas
o cuya evolución cursa a brotes. -No se puede aplicar cuando no es posible
asegurar la desaparición el efecto de la primera intervención en todos los
participantes. -Cualquier pérdida durante el seguimiento
tendrá mayores repercusiones, ya que cada paciente aporta mayor cantidad de información. |
CONSIDERACIONES FINALES
Rara vez un estudio puede
ser considerado muy bueno o muy malo en sí mismo. La valoración solo tiene
sentido en relación con la aportación que hace a lo que ya se sabe sobre la
cuestión.
No basta con juzgar un estudio por su metodología,
sino que los resultados deben contextualizarse en referencia:
-
a las probabilidades prácticas de probar determinadas hipótesis, y rara
vez es factible el ideal.
-
en estrecha relación con los conocimientos de los que las hipótesis
emergen.
Con frecuencia la pregunta
es: Teniendo en cuenta lo que ya se sabe sobre el tema ¿deberíamos complicarnos
más la vida, o sea el estudio?. De la repuesta dependerá que los estudios sean
más bien relevantes o manifiestamente fútiles.
BIBLIOGRAFíA RECOMENDADA
1. Clinical Trials. En Altman
(Ed.) Practical statistics for medical research. DG Chapman and Hall, Londres
10993;: 441-476
2.
Armitage P. The role of randomisation in clinical trials. Statistics in
Medicine 1982; 1: 345-352
3.
Friedman IM, Furberg CD, De Mets DL. Fundamentals of Clinical Trials. Boston.
John Wright. 1983
4.
Lavoriu P, Louis TA, Bailar JC, Polansky M. Designs for experiments. Parallel
comparison of treatment. N Eng J Med 1993; 309: 1291-1299
5.
Shapiro SH, Louis TA. Clinical Trials. Issues and approaches. Nueva York. Marcel
Dekker . 1996